
Mitochondrion
Background to the schools Wikipedia
The articles in this Schools selection have been arranged by curriculum topic thanks to SOS Children volunteers. See http://www.soschildren.org/sponsor-a-child to find out about child sponsorship.

In cell biology, a mitochondrion (plural mitochondria) is a membrane-enclosed organelle found in most eukaryotic cells. These organelles range from 1–10 micrometers ( μm) in size. Mitochondria are sometimes described as "cellular power plants" because they generate most of the cell's supply of adenosine triphosphate (ATP), used as a source of chemical energy. In addition to supplying cellular energy, mitochondria are involved in a range of other processes, such as signaling, cellular differentiation, cell death, as well as the control of the cell cycle and cell growth. Mitochondria have been implicated in several human diseases, including mental disorders, cardiac dysfunction, and may play a role in the aging process. The word mitochondrion comes from the Greek μίτος or mitos, thread + χονδρίον or khondrion, granule. Their ancestry is not fully understood, but, according to the endosymbiotic theory, mitochondria are descended from ancient bacteria, which were engulfed by the ancestors of eukaryotic cells more than a billion years ago.
Several characteristics make mitochondria unique. The number of mitochondria in a cell varies widely by organism and tissue type. Many cells have only a single mitochondrion, whereas others can contain several thousand mitochondria. The organelle is composed of compartments that carry out specialized functions. These compartments or regions include the outer membrane, the intermembrane space, the inner membrane, and the cristae and matrix. Mitochondrial proteins vary depending on the tissues and species. In human, 615 distinct types of proteins were identified from cardiac mitochondria; whereas in murine, 940 proteins encoded by distinct genes were reported. Mitochondrial proteome is thought to be dynamically regulated. Although most of a cell's DNA is contained in the cell nucleus, the mitochondrion has its own independent genome. Further, its DNA shows substantial similarity to bacterial genomes.
Structure
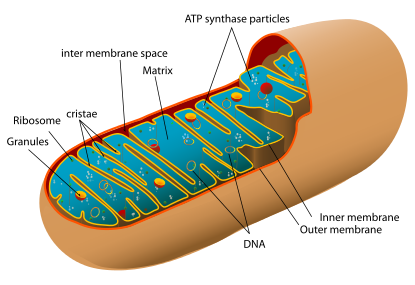
A mitochondrion contains inner and outer membranes composed of phospholipid bilayers and proteins. The two membranes, however, have different properties. Because of this double-membraned organization, there are five distinct compartments within the mitochondrion. There is the outer mitochondrial membrane, the intermembrane space (the space between the outer and inner membranes), the inner mitochondrial membrane, the cristae space (formed by infoldings of the inner membrane), and the matrix (space within the inner membrane).
Outer membrane
The outer mitochondrial membrane, which encloses the entire organelle, has a protein-to- phospholipid ratio similar to that of the eukaryotic plasma membrane (about 1:1 by weight). It contains large numbers of integral proteins called porins. These porins form channels that allow molecules 5000 Daltons or less in molecular weight to freely diffuse from one side of the membrane to the other. Larger proteins can also enter the mitochondrion if a signaling sequence at their N-terminus binds to a large multisubunit protein called translocase of the outer membrane, which then actively moves them across the membrane. Disruption of the outer membrane permits proteins in the intermembrane space to leak into the cytosol, leading to certain cell death.
Intermembrane space
The intermembrane space is the space between the outer membrane and the inner membrane. Because the outer membrane is freely permeable to small molecules, the concentrations of small molecules such as ions and sugars in the intermembrane space is the same as the cytosol. However, as large proteins must have a specific signaling sequence to be transported across the outer membrane, the protein composition of this space is different than the protein composition of the cytosol. One protein that is localized to the intermembrane space in this way is cytochrome c.
Inner membrane
The inner mitochondrial membrane contains proteins with four types of functions:
- Those that perform the redox reactions of oxidative phosphorylation
- ATP synthase, which generates ATP in the matrix
- Specific transport proteins that regulate metabolite passage into and out of the matrix
- Protein import machinery.
It contains more than 100 different polypeptides, and has a very high protein-to-phospholipid ratio (more than 3:1 by weight, which is about 1 protein for 15 phospholipids). The inner membrane is home to around 1/5 of the total protein in a mitochondrion. In addition, the inner membrane is rich in an unusual phospholipid, cardiolipin. This phospholipid was originally discovered in beef hearts in 1942, and is usually characteristic of mitochondrial and bacterial plasma membranes. Cardiolipin contains four fatty acids rather than two and may help to make the inner membrane impermeable. Unlike the outer membrane, the inner membrane does not contain porins and is highly impermeable to all molecules. Almost all ions and molecules require special membrane transporters to enter or exit the matrix. Proteins are ferried into the matrix via the translocase of the inner membrane (TIM) complex or via Oxa1. In addition, there is a membrane potential across the inner membrane formed by the action of the enzymes of the electron transport chain.
Cristae

The inner mitochondrial membrane is compartmentalized into numerous cristae, which expand the surface area of the inner mitochondrial membrane, enhancing its ability to produce ATP. These are not simple random folds but rather invaginations of the inner membrane, which can affect overall chemiosmotic function. In typical liver mitochondria, for example, the surface area, including cristae, is about five times that of the outer membrane. Mitochondria of cells that have greater demand for ATP, such as muscle cells, contain more cristae than typical liver mitochondria.
Matrix
The matrix is the space enclosed by the inner membrane. It contains about 2/3 of the total protein in a mitochondrion. The matrix is important in the production of ATP with the aid of the ATP synthase contained in the inner membrane. The matrix contains a highly-concentrated mixture of hundreds of enzymes, special mitochondrial ribosomes, tRNA, and several copies of the mitochondrial DNA genome. Of the enzymes, the major functions include oxidation of pyruvate and fatty acids, and the citric acid cycle.
Mitochondria have their own genetic material, and the machinery to manufacture their own RNAs and proteins (see: protein biosynthesis). A published human mitochondrial DNA sequence revealed 16,569 base pairs encoding 37 total genes, 24 tRNA and rRNA genes and 13 peptide genes. The 13 mitochondrial peptides in humans are integrated into the inner mitochondrial membrane, along with proteins encoded by genes that reside in the host cell's nucleus.
Organization and distribution
Mitochondria are found in nearly all eukaryotes. They vary in number and location according to cell type. Substantial numbers of mitochondria are in the liver, with about 1000–2000 mitochondria per cell making up 1/5th of the cell volume. The mitochondria can be found nestled between myofibrils of muscle or wrapped around the sperm flagellum. Often they form a complex 3D branching network inside the cell with the cytoskeleton. The association with the cytoskeleton determines mitochondrial shape, which can affect the function as well. Recent evidence suggests vimentin, one of the components of the cytoskeleton, is critical to the association with the cytoskeleton.
Function
The most prominent roles of the mitochondrion are its production of ATP and regulation of cellular metabolism. The central set of reactions involved in ATP production are collectively known as the citric acid cycle. However, the mitochondrion has many other functions in addition to the production of ATP.
Energy conversion
A dominant role for the mitochondria is the production of ATP, as reflected by the large number of proteins in the inner membrane for this task. This is done by oxidizing the major products of glucose, pyruvate, and NADH, which are produced in the cytosol. This process of cellular respiration, also known as aerobic respiration, is dependent on the presence of oxygen. When oxygen is limited, the glycolytic products will be metabolized by anaerobic respiration, a process that is independent of the mitochondria. The production of ATP from glucose has an approximately 13-fold higher yield during aerobic respiration compared to anaerobic respiration.
Pyruvate: the citric acid cycle
Each pyruvate molecule produced by glycolysis is actively transported across the inner mitochondrial membrane, and into the matrix where it is oxidized and combined with coenzyme A to form CO2, acetyl-CoA, and NADH.
The acetyl-CoA is the primary substrate to enter the citric acid cycle, also known as the tricarboxylic acid (TCA) cycle or Krebs cycle. The enzymes of the citric acid cycle are located in the mitochondrial matrix, with the exception of succinate dehydrogenase, which is bound to the inner mitochondrial membrane as part of Complex II. The citric acid cycle oxidizes the acetyl-CoA to carbon dioxide, and, in the process, produces reduced cofactors (three molecules of NADH and one molecule of FADH2) that are a source of electrons for the electron transport chain, and a molecule of GTP (that is readily converted to an ATP).
NADH and FADH2: the electron transport chain

(1) nucleolus
(2) nucleus
(3) ribosomes (little dots)
(4) vesicle
(5) rough endoplasmic reticulum (ER)
(6) Golgi apparatus
(7) Cytoskeleton
(8) smooth ER
(9) mitochondria
(10) vacuole
(11) cytoplasm
(12) lysosome
(13) centrioles within centrosome
The redox energy from NADH and FADH2 is transferred to oxygen (O2) in several steps via the electron transport chain. These energy-rich molecules are produced within the matrix via the citric acid cycle but are also produced in the cytoplasm by glycolysis. Reducing equivalents from the cytoplasm can be imported via the malate-aspartate shuttle system of antiporter proteins or feed into the electron transport chain using a glycerol phosphate shuttle. Protein complexes in the inner membrane ( NADH dehydrogenase, cytochrome c reductase, and cytochrome c oxidase) perform the transfer and the incremental release of energy is used to pump protons (H+) into the intermembrane space. This process is efficient, but a small percentage of electrons may prematurely reduce oxygen, forming reactive oxygen species such as superoxide. This can cause oxidative stress in the mitochondria and may contribute to the decline in mitochondrial function associated with the aging process.
As the proton concentration increases in the intermembrane space, a strong electrochemical gradient is established across the inner membrane. The protons can return to the matrix through the ATP synthase complex, and their potential energy is used to synthesize ATP from ADP and inorganic phosphate (Pi). This process is called chemiosmosis, and was first described by Peter Mitchell who was awarded the 1978 Nobel Prize in Chemistry for his work. Later, part of the 1997 Nobel Prize in Chemistry was awarded to Paul D. Boyer and John E. Walker for their clarification of the working mechanism of ATP synthase.
Heat production
Under certain conditions, protons can re-enter the mitochondrial matrix without contributing to ATP synthesis. This process is known as proton leak or mitochondrial uncoupling and is due to the facilitated diffusion of protons into the matrix. The process results in the unharnessed potential energy of the proton electrochemical gradient being released as heat. The process is mediated by a proton channel called thermogenin, or UCP1. Thermogenin is a 33k Da protein first discovered in 1973. Thermogenin is primarily found in brown adipose tissue, or brown fat, and is responsible for non-shivering thermogenesis. Brown adipose tissue is found in mammals, and is at its highest levels in early life and in hibernating animals. In humans, brown adipose tissue is present at birth and decreases with age.
Storage of calcium ions
The concentrations of free calcium in the cell can regulate an array of reactions and is important for signal transduction in the cell. Mitochondria can transiently store calcium, a contributing process for the cell's homeostasis of calcium. In fact, their ability to rapidly take in calcium for later release makes them very good "cytosolic buffers" for calcium. The endoplasmic reticulum (ER) is the most significant storage site of calcium, and there is a significant interplay between the mitochondrion and ER with regard to calcium. The calcium is taken up into the matrix by a calcium uniporter on the inner mitochondrial membrane. It is primarily driven by the mitochondrial membrane potential. Release of this calcium back into the cell's interior can occur via a sodium-calcium exchange protein or via "calcium-induced-calcium-release" pathways. This can initiate calcium spikes or calcium waves with large changes in the membrane potential. These can activate a series of second messenger system proteins that can coordinate processes such as neurotransmitter release in nerve cells and release of hormones in endocrine cells.
Additional functions
Mitochondria play a central role in many other metabolic tasks, such as:
- Regulation of the membrane potential
- Apoptosis-programmed cell death
- Glutamate-mediated excitotoxic neuronal injury
- Cellular proliferation regulation
- Regulation of cellular metabolism
- Certain heme synthesis reactions (see also: porphyrin)
- Steroid synthesis.
Some mitochondrial functions are performed only in specific types of cells. For example, mitochondria in liver cells contain enzymes that allow them to detoxify ammonia, a waste product of protein metabolism. A mutation in the genes regulating any of these functions can result in mitochondrial diseases.
Origin
Mitochondria have many features in common with prokaryotes. As a result, they are believed to be originally derived from endosymbiotic prokaryotes.
A mitochondrion contains DNA, which is organized as several copies of a single, circular chromosome. This mitochondrial chromosome contains genes for ribosomes, and the twenty-one tRNA's necessary for the translation of messenger RNAs into protein. The circular structure is also found in prokaryotes, and the similarity is extended by the fact that mitochondrial DNA is organized with a variant genetic code similar to that of Proteobacteria. This suggests that their ancestor, the so-called proto-mitochondrion, was a member of the Proteobacteria. In particular, the proto-mitochondrion was probably related to the rickettsia. However, the exact relationship of the ancestor of mitochondria to the alpha-proteobacteria and whether the mitochondria was formed at the same time or after the nucleus, remains controversial.
The ribosomes coded for by the mitochondrial DNA are similar to those from bacteria in size and structure. They closely resemble the bacterial 70S ribosome and not the 80S cytoplasmic ribosomes which are coded for by nuclear DNA.
The endosymbiotic relationship of mitochondria with their host cells was popularized by Lynn Margulis. The endosymbiotic hypothesis suggests that mitochondria descended from bacteria that somehow survived endocytosis by another cell, and became incorporated into the cytoplasm. The ability of these bacteria to conduct respiration in host cells that had relied on glycolysis and fermentation would have provided a considerable evolutionary advantage. In a similar manner, host cells with symbiotic bacteria capable of photosynthesis would also have had an advantage. The incorporation of symbiotes would have increased the number of environments in which the cells could survive. This symbiotic relationship probably developed 1.7-2 billion years ago.
A few groups of unicellular eukaryotes lack mitochondria: the microsporidians, metamonads, and archamoebae. These groups appear as the most primitive eukaryotes on phylogenetic trees constructed using rRNA information, suggesting that they appeared before the origin of mitochondria. However, this is now known to be an artifact of long-branch attraction – they are derived groups and retain genes or organelles derived from mitochondria (e.g., mitosomes and hydrogenosomes).
Genome
The human mitochondrial genome is a circular DNA molecule of about 16 kilobases. It encodes 37 genes: 13 for subunits of respiratory complexes I, III, IV, and V, 22 for mitochondrial tRNA, and 2 for rRNA. One mitochondrion can contain two to ten copies of its DNA.
As in prokaryotes, there is a very high proportion of coding DNA and an absence of repeats. Mitochondrial genes are transcribed as multigenic transcripts, which are cleaved and polyadenylated to yield mature mRNAs. Not all proteins necessary for mitochondrial function are encoded by the mitochondrial genome; most are coded by genes in the cell nucleus and the corresponding proteins imported into the mitochondrion. The exact number of genes encoded by the nucleus and the mitochondrial genome differs between species. In general, mitochondrial genomes are circular, although exceptions have been reported. Also, in general, mitochondrial DNA lacks introns, as is the case in the human mitochondrial genome; however, introns have been observed in some eukaryotic mitochondrial DNA, such as that of yeast and protists, including Dictyostelium discoideum.
While slight variations on the standard code had been predicted earlier, none was discovered until 1979, when researchers studying human mitochondrial genes determined that they used an alternative code. Many slight variants have been discovered since, including various alternative mitochondrial codes. Further, the AUA, AUC, and AUU codons are all allowable start codons.
| Organism | Codon | Standard | Novel |
|---|---|---|---|
| Mammalian | AGA, AGG | Arginine | Stop codon |
| AUA | Isoleucine | Methionine | |
| UGA | Stop codon | Tryptophan | |
| Invertebrates | AGA, AGG | Arginine | Serine |
| AUA | Isoleucine | Methionine | |
| UGA | Stop codon | Tryptophan | |
| Yeast | AUA | Isoleucine | Methionine |
| UGA | Stop codon | Tryptophan | |
| CUA | Leucine | Threonine |
Some of these differences should be regarded as pseudo-changes in the genetic code due to the phenomenon of RNA editing, which is common in mitochondria. In higher plants, it was thought that CGG encoded for tryptophan and not arginine; however, the codon in the processed RNA was discovered to be the UGG codon, consistent with the universal genetic code for tryptophan. Of note, the arthropod mitochondrial genetic code has undergone parallel evolution within a phylum, with some organisms uniquely translating AGG to lysine.
Mitochondrial genomes have far fewer genes than the eubacteria from which they are thought to be descended. Although some have been lost altogether, many have been transferred to the nucleus, such as the respiratory complex II protein subunits. This is thought to be relatively common over evolutionary time. A few organisms, such as the Cryptosporidium, actually have mitochondria that lack any DNA, presumably because all their genes have been lost or transferred. In Cryptosporidium, the mitochondria have an altered ATP generation system that renders the parasite resistant to many classical mitochondrial inhibitors such as cyanide, azide, and atovaquone.
Replication and inheritance
Mitochondria divide by binary fission similar to bacterial cell division; unlike bacteria, however, mitochondria can also fuse with other mitochondria.. The regulation of this division differs between eukaryotes. In many single-celled eukaryotes, their growth and division is linked to the cell cycle. For example, a single mitochondrion may divide synchronously with the nucleus. This division and segregation process must be tightly controlled so that each daughter cell receives at least one mitochondrion. In other eukaryotes (in humans for example), mitochondria may replicate their DNA and divide mainly in response to the energy needs of the cell, rather than in phase with the cell cycle. When the energy needs of a cell are high, mitochondria grow and divide. When the energy use is low, mitochondria are destroyed or become inactive. In such examples, and in contrast to the situation in many single celled eukaryotes, mitochondria are apparently randomly distributed to the daughter cells during the division of the cytoplasm.
An individual's mitochondrial genes are not inherited by the same mechanism as nuclear genes. At fertilization of an egg cell by a sperm, the egg nucleus and sperm nucleus each contribute equally to the genetic makeup of the zygote nucleus. In contrast, the mitochondria, and therefore the mitochondrial DNA, usually comes from the egg only. The sperm's mitochondria enter the egg but does not contribute genetic information to the embryo. Instead, paternal mitochondria are marked with ubiquitin to select them for later destruction inside the embryo. The egg cell contains relatively few mitochondria, but it is these mitochondria that survive and divide to populate the cells of the adult organism. Mitochondria are, therefore, in most cases inherited down the female line, known as maternal inheritance. This mode is seen in most organisms including all animals. However, mitochondria in some species can sometimes be inherited paternally. This is the norm among certain coniferous plants, although not in pine trees and yew trees. It has also been suggested that it occurs at a very low level in humans.
Uniparental inheritance leads to little opportunity for genetic recombination between different lineages of mitochondria, although a single mitochondrion can contain 2–10 copies of its DNA. For this reason, mitochondrial DNA usually is thought to reproduce by binary fission. What recombination does take place maintains genetic integrity rather than maintaining diversity. However, there are studies showing evidence of recombination in mitochondrial DNA. It is clear that the enzymes necessary for recombination are present in mammalian cells. Further, evidence suggests that animal mitochondria can undergo recombination. The data are a bit more controversial in humans, although indirect evidence of recombination exists. If recombination does not occur, the whole mitochondrial DNA sequence represents a single haplotype, which makes it useful for studying the evolutionary history of populations.
Population genetic studies
The near-absence of genetic recombination in mitochondrial DNA makes it a useful source of information for scientists involved in population genetics and evolutionary biology. Because all the mitochondrial DNA is inherited as a single unit, or haplotype, the relationships between mitochondrial DNA from different individuals can be represented as a gene tree. Patterns in these gene trees can be used to infer the evolutionary history of populations. The classic example of this is in human evolutionary genetics, where the molecular clock can be used to provide a recent date for mitochondrial Eve. This is often interpreted as strong support for a recent modern human expansion out of Africa. Another human example is the sequencing of mitochondrial DNA from Neanderthal bones. The relatively-large evolutionary distance between the mitochondrial DNA sequences of Neanderthals and living humans has been interpreted as evidence for lack of interbreeding between Neanderthals and anatomically-modern humans.
However, mitochondrial DNA reflects the history of only females in a population and so may not represent the history of the population as a whole. This can be partially overcome by the use of paternal genetic sequences, such as the non-recombining region of the Y-chromosome. In a broader sense, only studies that also include nuclear DNA can provide a comprehensive evolutionary history of a population.
Dysfunction and disease
Mitochondrial diseases
With their central place in cell metabolism, damage - and subsequent dysfunction - in mitochondria is an important factor in a wide range of human diseases. Mitochondrial disorders often present as neurological disorders, but can manifest as myopathy, diabetes, multiple endocrinopathy, or a variety of other systemic manifestations. Diseases caused by mutation in the mtDNA include Kearns-Sayre syndrome, MELAS syndrome and Leber's hereditary optic neuropathy. In the vast majority of cases, these diseases are transmitted by a female to her children, as the zygote derives its mitochondria and hence its mtDNA from the ovum. Diseases such as Kearns-Sayre syndrome, Pearson's syndrome, and progressive external ophthalmoplegia are thought to be due to large-scale mtDNA rearrangements, whereas other diseases such as MELAS syndrome, Leber's hereditary optic neuropathy, myoclonic epilepsy with ragged red fibers (MERRF), and others are due to point mutations in mtDNA.
In other diseases, defects in nuclear genes lead to dysfunction of mitochondrial proteins. This is the case in Friedreich's ataxia, hereditary spastic paraplegia, and Wilson's disease. These diseases are inherited in a dominance relationship, as applies to most other genetic diseases. A variety of disorders can be caused by nuclear mutations of oxidative phosphorylation enzymes, such as coenzyme Q10 deficiency and Barth syndrome. Environmental influences may also interact with hereditary predispositions and cause mitochondrial disease. For example, there may be a link between pesticide exposure and the later onset of Parkinson's disease.
Other diseases not directly linked to mitochondrial enzymes may feature dysfunction of mitochondria. These include schizophrenia, bipolar disorder, dementia, Alzheimer's disease, Parkinson's disease, epilepsy, stroke, cardiovascular disease, retinitis pigmentosa, and diabetes mellitus. The common thread linking these seemingly-unrelated conditions is cellular damage causing oxidative stress and the accumulation of reactive oxygen species. These oxidants then damage the mitochondrial DNA, resulting in mitochondrial dysfunction and cell death.
Possible relationships to aging
Given the role of mitochondria as the cell's powerhouse, there may be some leakage of the high-energy electrons in the respiratory chain to form reactive oxygen species. This can result in significant oxidative stress in the mitochondria with high mutation rates of mitochondrial DNA. A vicious cycle is thought to occur, as oxidative stress leads to mitochondrial DNA mutations, which can lead to enzymatic abnormalities and further oxidative stress. A number of changes occur to mitochondria during the aging process. Tissues from elderly patients show a decrease in enzymatic activity of the proteins of the respiratory chain. Large deletions in the mitochondrial genome can lead to high levels of oxidative stress and neuronal death in Parkinson's disease. Hypothesized links between aging and oxidative stress are not new and were proposed over 50 years ago; however, there is much debate over whether mitochondrial changes are causes of aging or merely characteristics of aging. One notable study in mice demonstrated no increase in reactive oxygen species despite increasing mitochondrial DNA mutations, suggesting that the aging process is not due to oxidative stress. As a result, the exact relationships between mitochondria, oxidative stress, and aging have not yet been settled.